Research
Our research group aims to discover, develop and understand new transition metal-catalyzed reactions. The scope of our studies encompasses the functionalization of C–H bonds in substrates ranging from alkanes to complex natural products and polymers, the enantioselective formation of C–C, C–N and C–O bonds by additions to alkenes and allylic substitution, and the creation of artificial metalloenzymes for applications in organic synthesis.
Regioselective Functionalization of Alkyl and Aryl C–H Bonds

Our group has discovered some of the most widely practiced C–H bond functionalization reactions. We discovered the reactions of metal boryl compounds with arenes and alkanes to form functionalized products and developed this new elementary reaction into catalytic processes for the conversion of alkyl and aryl C–H bonds into C–B bonds. The C–B bonds in the product can then be converted to C–C and C–heteroatom bonds in a wide range of valuable products.

Our success on the functionalization of C–H bonds in complex molecules with boranes and silanes has led us to develop additional classes of C–H bond functionalizations. For example, we recently disclosed a combination of catalyst and reagent for the conversion of C–H bonds in natural products to the C–N bonds in alkyl azides.
Olefin Hydrofunctionalization

The hydrofunctionalization of olefins is a long-standing goal for transition metal catalysis, and we reported the first additions of N–H and O–H bonds to unactivated alkenes with transition metal catalysts. More recently, we discovered the first anti-Markovnikov addition of an unactivated arene to an unactivated alkene and the first additions of an N–H bond to internal alkenes, including enantioselective versions of this hydroamination reaction.

Enantioselective Allylic Amination and Etherification

We have developed an iridium catalyst that forms chiral allylic amines, ethers, and carbonyl compounds in high enantiomeric excess from terminal allylic esters. Detailed mechanistic studies have shown that the catalyst forms by cyclometalation at a methyl C–H bond of the ligand, and this observation has allowed the design of simplified, as well as improved catalysts.
New Classes of Cross-Couplings

We developed a palladium-catalyzed process that forms arylamines, aryl sulfides, and arylethers. This reaction has become one of the most widely practiced reactions by medicinal chemists discovered in the past four decades. The catalytic chemistry resulted from our detailed mechanistic experiments on transition metal amide, alkoxo and thiolato complexes.

We have developed a simple method to convert aryl halides and ketones, esters, amides, cyanoesters, malonates, nitriles, and related compounds to alpha aryl carbonyl compounds and nitriles in the presence of base and a palladium catalyst. Familiar compounds that can be generated from these products include Ibuprofen, Naproxin and Tamoxifen.
Metal Catalyzed Fluorination and Fluoroalkylation
The synthesis of arenes and heteroarenes containing fluorine is crucial to the discovery of modern drugs and agrochemicals. These fluorine atoms impart a unique combination of steric and electronic properties and can influence the interactions of the molecules with proteins by non-covalent interactions.



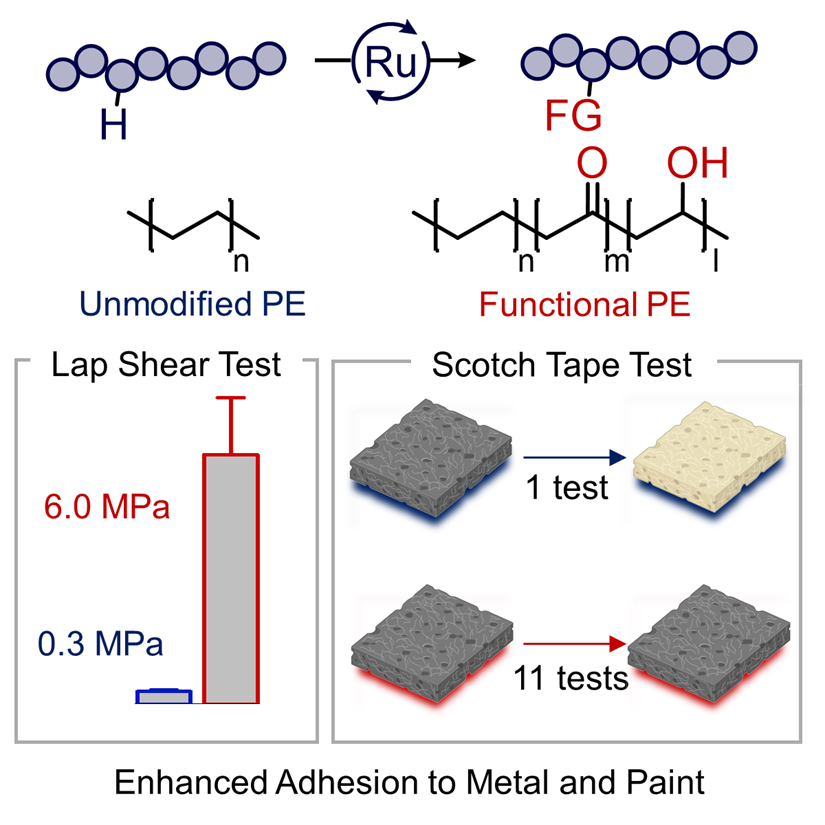
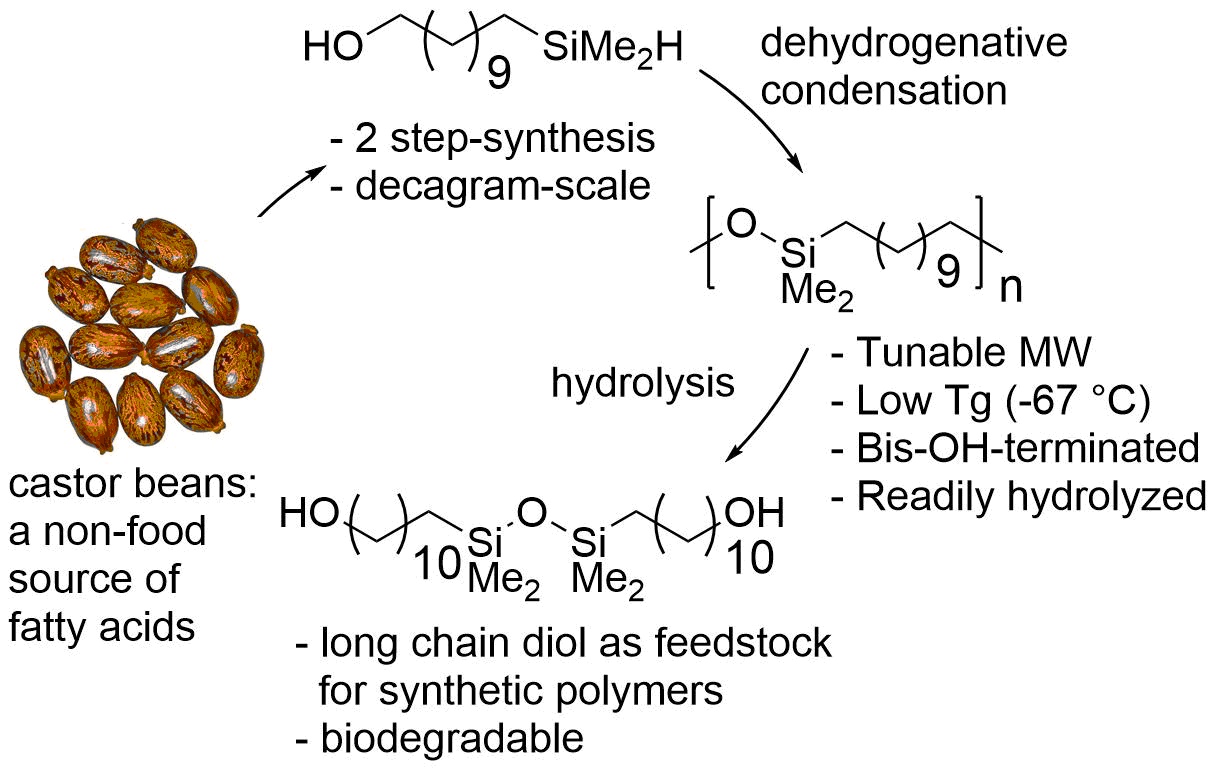
Catalysis for Renewable Chemicals: Catalysis for the Upcycling of Polymers
The world manufactures approximately 300 billion pounds of polyolefins a year, and little of this plastic is recycled. We envision a future in which such plastic is chemically converted to more valuable products, converted to polymers with a more cyclic lifetime, and even created from the outset to have a cyclic lifetime by design.
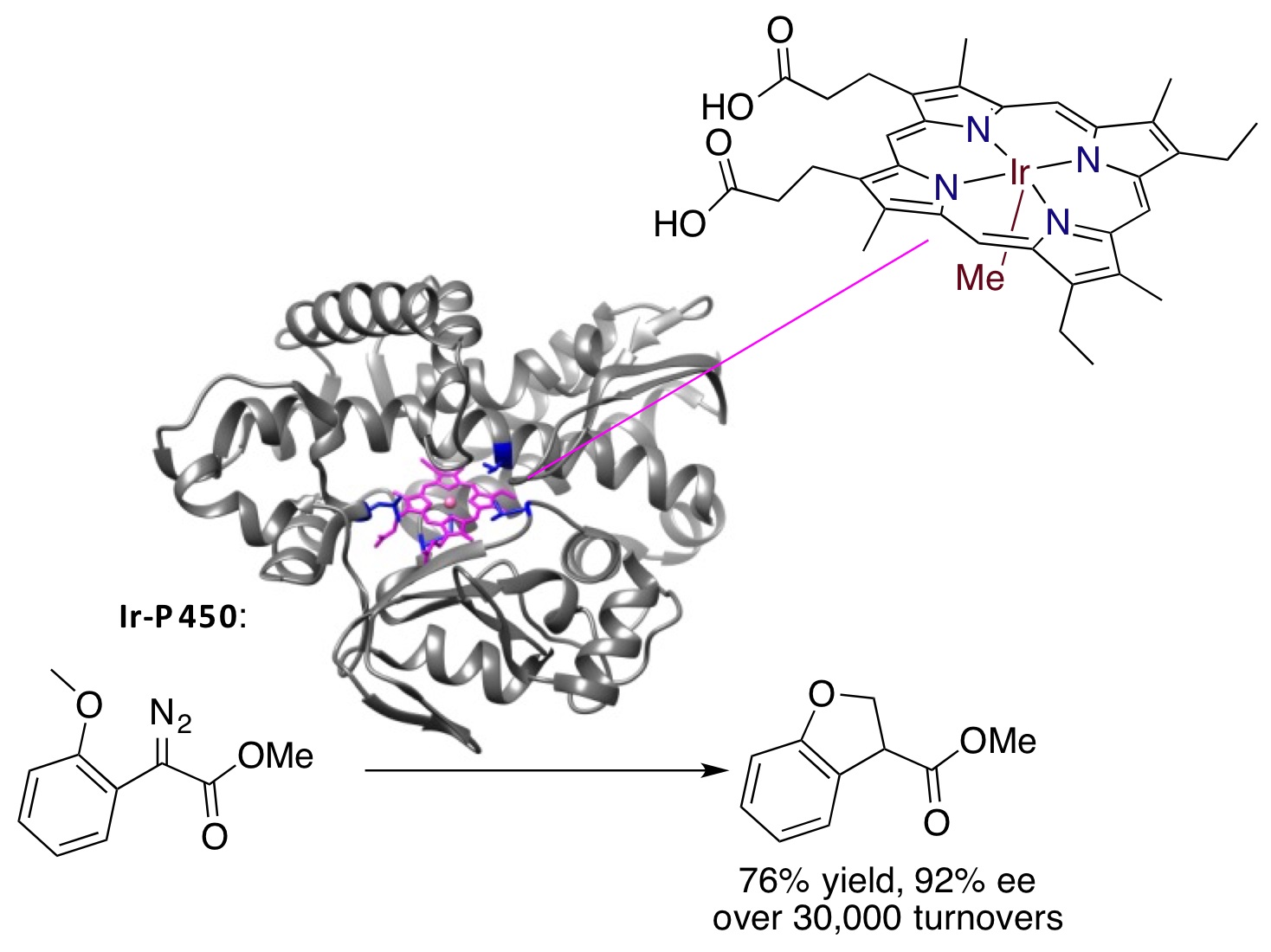
Artificial Metalloenzymes
In addition to studies with small-molecule catalysts, we have begun to develop methods to combine the diversity of reactions of organometallic complexes with the selectivity of enzymes. To do so, we have followed several approaches, including cooperative chemoenzymatic reactions and artificial metalloenzymes in which the active site contains an organometallic complex.


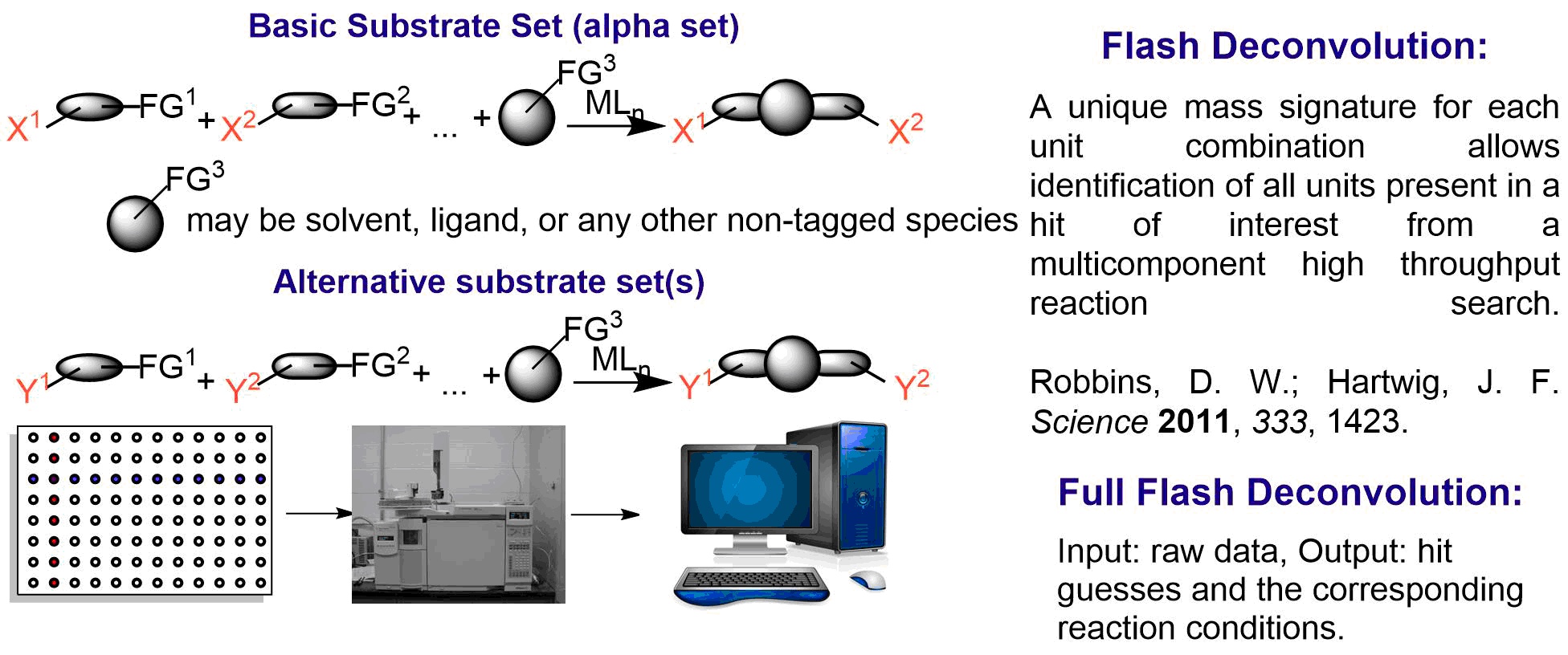
Accelerating Discovery using Machine Learning and Data Science
We have been developing alternative approaches to the conventional methods for catalyst discovery and development. In particular, we have used fluorescent and colorimetric assays to evaluate catalysts for carbon-carbon and carbon-nitrogen bond forming processes catalyzed by transition metal complexes. Recently, we are using machine learning approaches to predict catalyst activity and selectivity from large datasets.