Improved catalysts for the palladium-catalyzed synthesis of oxindoles by amide a-arylation. Rate acceleration, use of aryl chloride substrates, and a new carbene ligand for asymmetric transformations
J. Org. Chem., 2001, 66 (3402-3415)
View on publisher site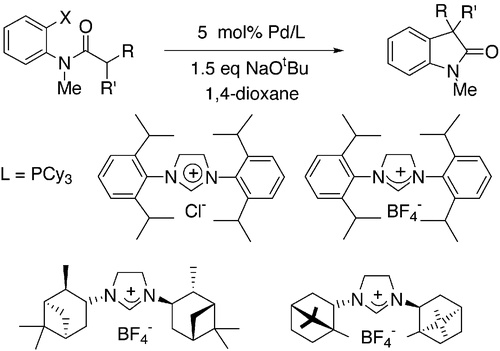
Abstract
Catalysts comprised Pd(OAc)2 and either PCy3 or sterically hindered N-heterocyclic carbene ligands provide fast rates for a palladium-catalyzed synthesis of oxindoles by amide α-arylation. This catalyst system allowed for room-temperature reactions in some cases and reactions of aryl chlorides at 70 °C. Most important, reactions occurred in high yields under mild conditions to form the quaternary carbon in α,α-disubstituted oxindoles. The combined inter- and intramolecular reaction afforded an efficient synthetic method for formation of α-aryloxindole derivatives. Surprisingly, catalysts containing tert-butylphosphine ligands, which have been most reactive for ketone arylations, were less active than those containing PCy3. Use of new, optically active heterocyclic carbene ligands gave substantial enantioselectivity in formation of an α,α-disubstituted oxindole. In contrast, a variety of optically active phosphine ligands that were tested gave poor enantioselectivity. Mechanistic studies showed that the reaction involves rate-limiting oxidative addition of aryl halide. Base-induced formation of and reductive elimination from an arylpalladium enolate intermediate were both faster than oxidative addition. Deprotonation of the tethered amide appeared to be faster than reductive elimination of the resulting palladium enolate to form the oxindole product.
Read on publisher's site