Direct Observation of Diastereomeric α-C-Bound Enolates during Enantioselective α-Arylations: Synthesis, Characterization, and Reactivity of Arylpalladium Fluorooxindole Complexes
J. Am. Chem. Soc., 2021, 143 (11741-11750)
View on publisher site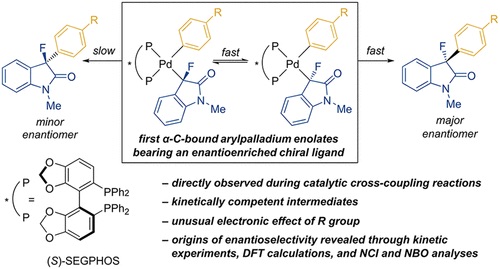
Abstract
The Pd-catalyzed asymmetric α-arylation of carbonyl compounds is a valuable strategy to form benzylic stereocenters. However, the origin of the stereoselectivity of these reactions is poorly understood, and little is known about the reactivity of the putative diastereomeric arylpalladium enolate intermediates. To this end, we report the synthesis and characterization of a series of diphosphine-ligated arylpalladium fluoroenolate complexes, including complexes bearing a metal-bound, stereogenic carbon and an enantioenriched chiral diphosphine ligand. These complexes reductively eliminate to form chiral α-aryl-α-fluorooxindoles with enantioselectivities and rates that are relevant to those of the catalytic process with SEGPHOS as the ancillary ligand. Kinetic studies showed that the rate of reductive elimination is slightly slower than the rate of epimerization of the intermediate, causing the reductive elimination step to impart the greatest influence on the enantioselectivity. DFT calculations of these processes are consistent with these experimental rates and suggest that the minor diastereomer forms the major enantiomer of the product. The rates of reductive elimination from complexes containing a variety of electronically varied aryl ligands revealed the unusual trend that complexes bearing more electron-rich aryl ligands react faster than those bearing more electron-poor aryl ligands. Noncovalent Interaction (NCI) and Natural Bond Orbital (NBO) analyses of the transition-state structures for reductive elimination from the SEGPHOS-ligated complexes revealed key donor–acceptor interactions between the Pd center and the fluoroenolate fragment. These interactions stabilize the pathway to the major product enantiomer more strongly than they stabilize that to the minor enantiomer.
Read on publisher's site