Iridium-Catalyzed, β-Selective C(sp3)-H Silylation of Aliphatic Amines to Form Silapyrrolidines and 1,2-Amino Alcohols
J. Am. Chem. Soc., 2018, 140 (18032-18038)
View on publisher site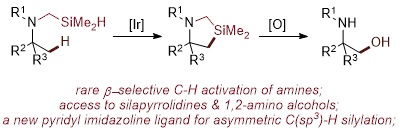
Abstract
The functionalization of unactivated C(sp3)-H bonds of aliphatic amines catalyzed by transition-metal complexes is an important because amine-based functionality is present in a majority of biologically active molecules and commercial pharmaceuticals. However, such reactions are under-developed and challenging to achieve in general because the basicity and reducing properties of alkylamines tends to interfere with potential reagents and catalysts. The functionalization of C-H bonds β to the nitrogen of aliphatic amines to form prevalent 1,2-amino functionalized structures is a particular challenge to achieve because the C-H bond b to nitrogen is stronger than the C-H bond α to nitrogen, and the nitrogen in the amine or its derivatives usually directs a catalyst to react at more distal γ- and δ-C-H bonds to form 5- or 6-membered metallacyclic intermediate. The enantioselective functionalization of a C-H bond at any position in amines also has been vexing and is currently limited to reactions of specific, sterically hindered, cyclic structures. We report iridium-catalyzed, β-selective silylations of unactivated C(sp3)-H bonds of aliphatic amines to form silapyrrolidines that are both silicon-containing analogs of common saturated nitrogen heterocycles and precursors to 1,2-amino alcohols by Tamao-Fleming oxidation. These silylations of amines are accomplished by introducing a simple methylene linker between the heteroatom and silicon that has not been used previously for the silylation of C-H bonds. The reactions occur with high enantioselectivity when catalyzed by complexes of new chiral, pyridyl imidazoline ligands, and the rates of reactions with catalysts of these highly basic ligands are particularly fast and even occur at or even below room temperature.
Read on publisher's site