Rhodium-Catalyzed Regioselective Silylation of Alkyl C–H Bonds for the Synthesis of 1,4-Diols
J. Am. Chem. Soc., 2018, 140 (1460-1470)
View on publisher site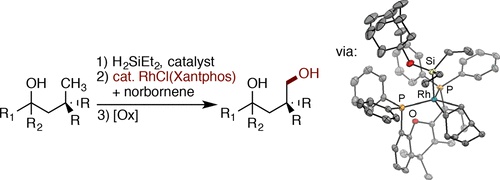
Abstract
A rhodium-catalyzed intramolecular silylation of alkyl C–H bonds has been developed that occurs with unusual selectivity for the C–H bonds located δ to the oxygen atom of an alcohol-derived silyl ether over typically more reactive C–H bonds more proximal to the same oxygen atom. (Hydrido)silyl ethers, generated in situ by dehydrogenative coupling of tertiary alcohols with diethylsilane, undergo regioselective silylation at a primary C–H bond δ to the hydroxyl group in the presence of [(Xantphos)Rh(Cl)] as catalyst. Oxidation of the resulting 6-membered oxasilolanes generates 1,4-diols. This silylation and oxidation sequence provides an efficient method to synthesize 1,4-diols by a hydroxyl-directed, aliphatic C–H bond functionalization reaction and is distinct from the synthesis of 1,3-diols from alcohols catalyzed by iridium. Mechanistic studies show that the rhodium-catalyzed silylation of alkyl C–H bonds occurs with a resting state and relative rates for elementary steps that are significantly different from those for the rhodium-catalyzed silylation of aryl C–H bonds. The resting state of the catalyst is a (Xantphos)Rh(I)(SiR3)(norbornene) complex, and an analogue was synthesized and characterized crystallographically. The rate-limiting step of the process is oxidative addition of the δ C–H bond to Rh. Computational studies elucidated the origin of high selectivity for silylation of the δ C–H bond when Xantphos-ligated rhodium is the catalyst. A high barrier for reductive elimination from the six-membered metalacyclic, secondary alkyl intermediate formed by cleavage of the γ C–H bond and low barrier for reductive elimination from the seven-membered metalacyclic, primary alkyl intermediate formed by cleavage of the δ C–H accounts for the selective functionalization of the δ C–H bond.
Read on publisher's site