Mechanistic Studies of Copper-Catalyzed Asymmetric Hydroboration of Alkenes
J. Am. Chem. Soc., 2017, 139 (12758-12772)
View on publisher site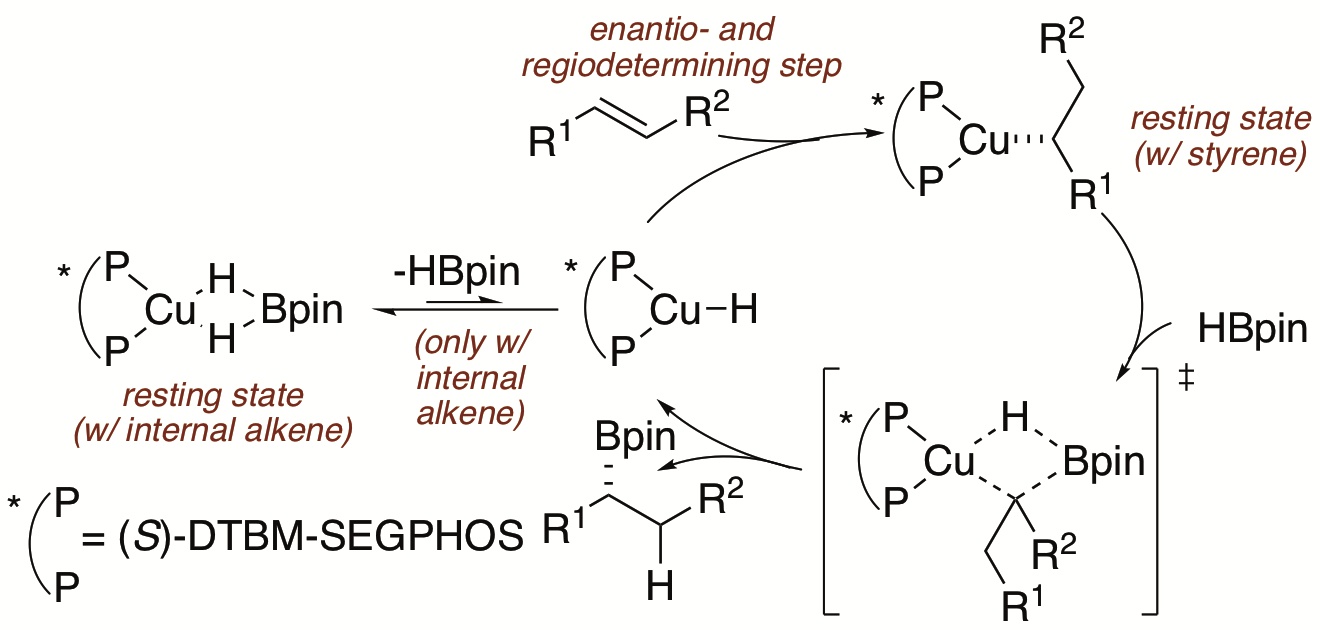
Abstract
Mechanistic studies of the copper-catalyzed asymmetric hydroboration of vinylarenes and internal alkenes are reported. Catalytic systems with both DTBM-SEGPHOS and SEGPHOS as the ligands have been investigated. With DTBM-SEGPHOS as the ligand, the resting state of the catalyst, which is also a catalytic intermediate, for hydroboration of 4-fluorostyrene is a phenethylcopper(I) complex ligated by the bisphosphine. This complex was fully characterized by NMR spectroscopy and x-ray crystallography. The turnover-limiting step in the catalytic cycle for the reaction of vinylarenes is the borylation of this phenethylcopper complex with pinacolborane (HBpin) to form the boronate ester product and a copper hydride. Experiments showed that the borylation occurs with retention of configuration at the benzylic position. β-Hydrogen elimination and insertion of the alkene to reform this phenethylcopper complex is reversible in the absence of HBpin but is irreversible during the catalytic process because reaction with HBpin is faster than β-hydrogen elimination of the phenethylcopper complex. Studies on the hydroboration of a representative internal alkene, trans-3-hexenyl 2,4,6-trichlorobenzoate, which undergoes enantioselective and regioselective addition of HBpin catalyzed by DTBM-SEGPHOS, KOtBu and CuCl, also was conducted, and these studies revealed that a DTBM-SEGPHOS-ligated copper(I) dihydridoborate complex is the resting state of the catalyst in this case. The turnover-limiting step in the catalytic cycle for hydroboration of the internal alkene is insertion of the alkene into a copper(I) hydride formed by reversible dissociation of HBpin from the copper dihydridoborate species. With SEGPHOS as the ligand, a dimeric copper hydride was observed as the dominant species during the hydroboration of 4-fluorostyrene, and this complex is not catalytically competent. DFT calculations provide a view into the origins of regioselectivity and enantioselectivity of the catalytic process and indicate that the charge on the copper-bound carbon and delocalization of charge onto the aryl ring controls the rate of the alkene insertion and the regioselectivity of the catalytic reactions of vinylarenes.
Read on publisher's site