Synthetic and Computational Studies on the Rhodium-Catalyzed Hydroamination of Aminoalkenes
ACS. Catal., 2016, 6 (5651-5665)
View on publisher site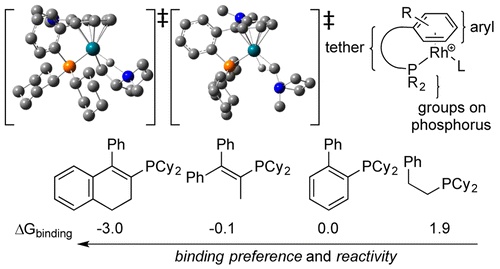
Abstract
The influence of ligand structure on rhodium-catalyzed hydroamination has been evaluated for a series of phosphinoarene ligands. These catalysts have been evaluated in a set of catalytic intramolecular Markovnikov hydroamination reactions. The mechanism of hydroamination catalyzed by the rhodium(I) complexes in this study was examined computationally, and the turnover-limiting step was elucidated. These computational studies were extended to a series of theoretical hydroamination catalysts to compare the electronic effects of the ancillary ligand substituents. The relative energies of intermediates and transition states were compared to those of intermediates in the reaction catalyzed by the unsubstituted catalyst. The experimental difference in the reactivities of electron-rich and electron-poor catalysts was compared to the computational results, and it was found that the activity for the electron-poor catalysts predicted from the reaction barriers was overestimated. Thus, the analysis of the catalysts in this study was expanded to include the binding preference of each ligand, in comparison to that of the unsubstituted ligand. This information accounts for the disparity between observed reactivity and the calculated overall reaction barrier for electron-poor ligands. The ligand-binding preferences for new ligand structures were calculated, and ligands that were predicted to bind strongly to rhodium generated catalysts for the experimental catalytic reactions that were more reactive than those predicted to bind more weakly.
Read on publisher's site