Assessment of the Electronic Factors Determining the Thermodynamics of "Oxidative Addition" of C–H and N–H Bonds to Ir(I) Complexes
J. Am. Chem. Soc., 2016, 138 (149-163)
View on publisher site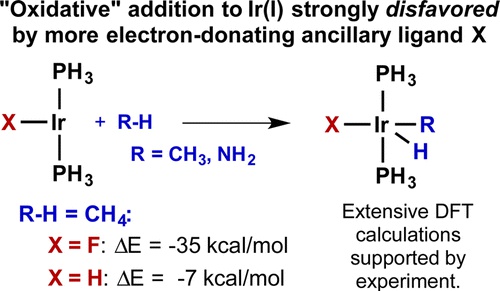
Abstract
A study of electronic factors governing the thermodynamics of C–H and N–H bond addition to Ir(I) complexes was conducted. DFT calculations were performed on an extensive series of trans-(PH3)2IrXL complexes (L = NH3 and CO; X = various monodentate ligands) to parametrize the relative σ- and π-donating/withdrawing properties of the various ligands, X. Computed energies of oxidative addition of methane to a series of three- and four-coordinate Ir(I) complexes bearing an ancillary ligand, X, were correlated with the resulting (σX, πX) parameter set. Regression analysis indicates that the thermodynamics of addition of methane to trans-(PH3)2IrX are generally strongly disfavored by increased σ-donation from the ligand X, in contradiction to widely held views on oxidative addition. The trend for oxidative addition of methane to four-coordinate Ir(I) was closely related to that observed for the three-coordinate complexes, albeit slightly more complicated. The computational analysis was found to be consistent with the rates of reductive elimination of benzene from a series of isoelectronic Ir(III) phenyl hydride complexes, measured experimentally in this work and previously reported. Extending the analysis of ancillary ligand energetic effects to the oxidative addition of ammonia to three-coordinate Ir(I) complexes leads to the conclusion that increasing σ-donation by X also disfavors oxidative addition of N–H bonds to trans-(PH3)2IrX. However, coordination of NH3 to the Ir(I) center is disfavored even more strongly by increasing σ-donation by X, which explains why the few documented examples of H–NH2 oxidative addition to transition metals involve complexes with strongly σ-donating ligands situated trans to the site of addition. An orbital-based rationale for the observed results is presented.
Read on publisher's site