Intramolecular Hydroamination of Unbiased and Functionalized Primary Aminoalkenes Catalyzed by a Rhodium Aminophosphine Complex
J. Am. Chem. Soc. , 2010, 132 (13813-13822)
View on publisher site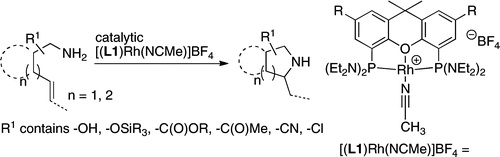
Abstract
We report a rhodium catalyst that exhibits high reactivity for the hydroamination of primary aminoalkenes that are unbiased toward cyclization and that possess functional groups incompatible with more electrophilic hydroamination catalysts. The rhodium catalyst contains an unusual diaminophosphine ligand (L1) that binds to rhodium in a κ3-P,O,P mode. The reactions catalyzed by this complex typically proceed at mild temperatures (room temperature to 70 °C) and occur with primary aminoalkenes lacking substituents on the alkyl chain that bias the system toward cyclization, with primary aminoalkenes containing chloride, ester, ether, enolizable ketone, nitrile, and unprotected alcohol functionality, and with primary aminoalkenes containing internal olefins. Mechanistic data imply that these reactions occur with a turnover-limiting step that is different from that of reactions catalyzed by late-transition-metal complexes of Pd, Pt, and Ir. This change in the turnover-limiting step and resulting high activity of the catalyst stem from favorable relative rates for protonolysis of the M−C bond to release the hydroamination product versus reversion of the aminoalkyl intermediate to regenerate the acyclic precursor. Probes of the origin of the reactivity of the rhodium complex of L1 imply that the aminophosphine groups lead to these favorable rates by effects beyond steric demands and simple electron donation to the metal center.
Read on publisher's site