Computational Studies on the Relative Rates for Migratory Insertions of Alkenes into Square-Planar Rhodium–Methyl, –Amide, and –Hydroxide Complexes
J. Am. Chem. Soc. , 2009, 131 (14703)
View on publisher site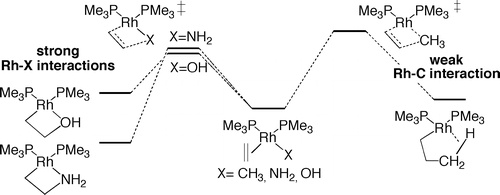
Abstract
The relative rates of the migratory insertions of alkenes into the M−X bonds of (PMe3)2Rh(η2-CH2═CHR)(X) (R = H, Me; X = CH3, NH2, OH) have been analyzed by DFT calculations. These insertions are computed to form metallacycles containing a metal−carbon bond and either an agostic interaction, a dative metal−nitrogen bond, or a dative metal−oxygen bond. The computed barriers for migratory insertion into the metal−hydroxo and metal−amido bonds are lower than those for insertion into the metal−methyl bond. Application of Bader’s atoms-in-molecules analysis and natural localized molecular orbital analysis implies that the barriers for alkene insertion into M−X bonds are controlled by the degree of M−X bonding in the transition state, which correlates with the degree of M−X bonding in the initial product. The Rh−X bond orders in the transition states for migratory insertion of ethylene into the Rh−NH2 and Rh−OH bonds in (PH3)2Rh(η2-C2H4)(NH2) and (PH3)2Rh(η2-C2H4)(OH) are much larger than that in the transition state for insertion into the Rh−C bond of (PH3)2Rh(η2-C2H4)(CH3). The free energy barriers for 1,2- and 2,1-insertions of propene into the rhodium complexes were also calculated, and the barrier for 1,2-insertion was found to be lower than that for 2,1-insertion. Most striking, the ΔΔG‡ values for 1,2- versus 2,1-insertion of propene into these rhodium complexes were calculated to increase in the order X = CH3 < NH2 < OH. The increasing stability of the 1,2-insertion product with increasing polarity of the C−X bonds parallels the relative stabilities of linear versus branched alkanes, amines, and alcohols.
Read on publisher's site