Mechanism of the Mild Functionalization of Arenes by Diboron Reagents Catalyzed by Iridium Complexes. Intermediacy and Chemistry of Bipyridine-Ligated Iridium Trisboryl Complexes
J. Am. Chem. Soc., 2005, 127 (14263-14278)
View on publisher site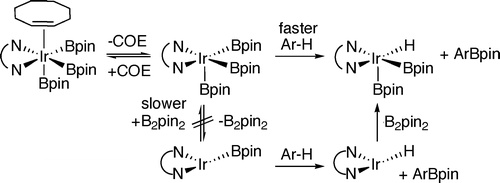
Abstract
This paper describes mechanistic studies on the functionalization of arenes with the diboron reagent B2pin2 (bis-pinacolato diborane(4)) catalyzed by the combination of 4,4‘-di-tert-butylbipyridine (dtbpy) and olefin-ligated iridium halide or olefin-ligated iridium alkoxide complexes. This work identifies the catalyst resting state as [Ir(dtbpy)(COE)(Bpin)3] (COE = cyclooctene, Bpin = 4,4,5,5-tetramethyl-1,3,2-dioxaborolanyl). [Ir(dtbpy)(COE)(Bpin)3] was prepared by independent synthesis in high yield from [Ir(COD)(OMe)]2, dtbpy, COE, and HBpin. This complex is formed in low yield from [Ir(COD)(OMe)]2, dtbpy, COE, and B2pin2. Kinetic studies show that this complex reacts with arenes after reversible dissociation of COE. An alternative mechanism in which the arene reacts with the Ir(I) complex [Ir(dtbpy)Bpin] after dissociation of COE and reductive elimination of B2pin2 does not occur to a measurable extent. The reaction of [Ir(dtbpy)(COE)(Bpin)3] with arenes and the catalytic reaction of B2pin2 with arenes catalyzed by [Ir(COD)(OMe)]2 and dtbpy occur faster with electron-poor arenes than with electron-rich arenes. However, both the stoichiometric and catalytic reactions also occur faster with the electron-rich heteroarenes thiophene and furan than with arenes, perhaps because η2-heteroarene complexes are more stable than the η2-arene complexes and the η2-heteroarene or arene complexes are intermediates that precede oxidative addition. Kinetic studies on the catalytic reaction show that [Ir(dtbpy)(COE)(Bpin)3] enters the catalytic cycle by dissociation of COE, and a comparison of the kinetic isotope effects of the catalytic and stoichiometric reactions shows that the reactive intermediate [Ir(dtbpy)(Bpin)3] cleaves the arene C−H bond. The barriers for ligand exchange and C−H activation allow an experimental assessment of several conclusions drawn from computational work. Most generally, our results corroborate the conclusion that C−H bond cleavage is turnover-limiting, but the experimental barrier for this bond cleavage is much lower than the calculated barrier.
Read on publisher's site