Iridium-Catalyzed Borylation of Primary Benzylic C–H Bonds without a Directing Group: Scope, Mechanism, and Origins of Selectivity
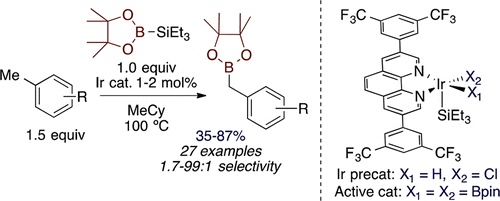
Primary benzylic boronate esters are useful intermediates in organic synthesis, but these reagents cannot be prepared by hydroboration. The benzylic C–H borylation of methylarenes would be a method to form these products, but such reactions without neat methylarene or a directing group are unknown. We report an approach to divert the borylation of methylarenes from aromatic positions to benzylic positions with a silylborane as reagent and a new iridium catalyst containing an electron-deficient phenanthroline as ligand. This system forms benzylic boronate esters selectively over the corresponding aryl boronate esters. An Ir diboryl monosilyl complex ligated by the phenanthroline was isolated and determined to be the resting state of the catalyst. Mechanistic studies show that this complex is kinetically competent to be an intermediate in the catalytic process. Kinetic studies of benzylic and aryl C–H borylation catalyzed by various Ir complexes show that the rate of aryl C–H borylation decreases with decreasing electron density at the metal center of the Ir catalyst, but that the rate of benzylic C–H borylation is less sensitive to the degree of electron density at the metal center of the Ir catalyst. Kinetic and computational studies suggest that the two borylation reactions respond differently to the degree of electron density at the metal center because they occur with different turnover-limiting steps. The turnover-limiting step in the borylation of aryl C–H bonds is known to be C–H oxidative addition, but the turnover-limiting step of the borylation of benzylic C–H bonds appears to be an isomerization prior to C–B reductive elimination.
Read more on publisher's site.