Catalysis with platinum-group alkylamido complexes, the active palladium amide in catalytic aryl halide aminations as deduced from kinetic data and independent generation
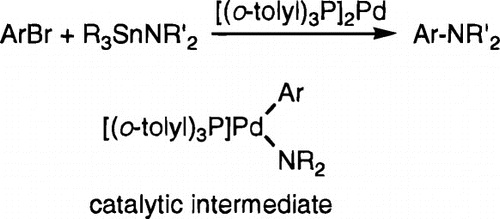
Mechanistic studies of the palladium-catalyzed coupling between aryl bromides and tin amides were conducted as a means to evaluate the pathway of this reaction as well as the general potential of low valent amido complexes to be reactive intermediates in catalysis. The specific systems involved reactions between Bu3SnNMe2 and aryl halides catalyzed by {Pd[P(o-Tol)3]2} (1), {Pd[P(o-Tol)3](p-MeC6H4)(Br)}2 (2a), and {Pd[P(o-Tol)3](NHMe2)(p-MeC6H4)(Br)} (3a). A combination of kinetic studies and independent synthesis of reaction intermediates indicated that the three-coordinate platinum-group amido complex {Pd[P(o-Tol)3](Ar)(NMe2)} was an intermediate in these reactions. Thus, these aryl halide aminations are rare examples of catalysis with a platinum-group amido complex. Kinetic data were obtained by 1H NMR spectroscopy, and the rate behavior was determined to be zero order in added phosphine, zero order in aryl halide, and first order in tin amide under conditions of equal or greater concentrations of aryl bromide compared to tin amide. Reactions catalyzed by 3a were first order in the palladium complex. Reaction rates were inhibited by added tin bromide, but not by the arylamine product. The inhibition by tin bromide showed that reversible transmetalation between an aryl halide complex and the tin reagent was occurring. Subsequent to reversible transmetalation, a rate-determining reductive elimination of arylamine occurred. Under conditions with a 10-fold excess of tin amide and high phosphine concentrations, the rate-determining-step became oxidative addition of aryl bromide, and reactions became first order, rather than zero order, in aryl bromide. The amido intermediate deduced from kinetic studies appeared to be generated by reacting {Pd[P(o-Tol)3](p-BuC6H4)(Br}2 (2b) with lithium arylamides or by deprotonating {Pd[P(o-Tol)3](NHEt2)(p-BuC6H4)(Br)} (3b) with MN(SiMe3)2 (M = K, Li). Both reactions gave yields of arylamine that were comparable to those of catalytic reactions. Competition and relative rate studies revealed an equilibrium between aryl halide complexes 2a−c and a tin amide adduct of it. In competition studies involving an in situ selectivity for reaction of Bu3SnNMe2 or Bu3SnNEt2 with p-t-BuC6H4Br catalyzed by 1, the ratio of N,N-dimethylaniline to N,N-diethylaniline was 2.9. However, kinetic measurements of individual reactions showed that Bu3SnNMe2 reacted only 1.4 times faster than Bu3SnNEt2, consistent with a reversible equilibrium involving tin amide binding to the catalyst, similar to that resulting from substrate binding preëquilibria in enzyme systems.
Read more on publisher's site.